
A recent New York Times article published on December 22, 2012 described efforts to develop new drug compounds capable of targeting cancer cells that arise in many different organs of the human body. Unfortunately, much was left unsaid with little source information to track down more specifics. Fierce Biotech regurgitated the essentials of the article, but little in the way of basic science was offered. Since this topic is of great interest here at CH3 BioSystems, some additional R&D context seems to be in order for the CH3 News web log.
The Times article suggests that the fatal liposarcoma that Mr. Bellino suffered from would have been an ideal test for a Sanofi drug developed to treat “…tumors that nearly always have the exact genetic problem the drug was meant to attack – a fusion of two large proteins.” Although Gina Kolata did not explicitly name the fused proteins, the description brings to mind the chimeric TLS-CHOP oncoprotein (Rabbitts et al., 1993) identified in malignant liposarcoma. TLS (Transformed in LipoSarcoma), known also as FUS (FUsed in Sarcoma), is an interesting methylarginine protein that is also implicated in the cause of a familial form of amyotrophic lateral sclerosis (Vance et al., 2009).
In 2011 a group in Japan presented data suggesting that the methylation of TLS activates transcription of survivin (Du et al., 2011), a member of the BIRC family of apoptosis inhibitory proteins that directly bind to and block caspases.
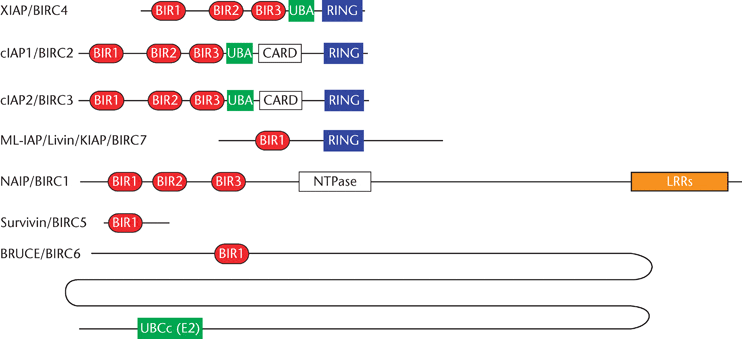
FIGURE 1. BIRC (Baculoviral Inhibitor of apoptosis Repeat Containing) Family of Apoptosis Inhibitory Proteins
So how are survivin and the arginine methylation of TLS biologically important for tumor growth? Why is this possibly relevant to fusion proteins, oncogenesis, protein-protein interactions, drug therapy and the article in the NY Times? Consider the series of facts below:
- Survivin expression is repressed by normal, wild type p53 (Mita et al., 2008)
- Mutation of p53 is frequently associated with solid tumors and is considered a likely reason for the abnormal apoptosis observed in tumors
- Thus, the cellular mechanism underlying p53 mutation-mediated tumor growth may be facilitated by aberrantly elevated levels of survivin expression
- Inactive p53 –> Elevated Survivin –> Inhibition of Apoptosis –> Increased tumor growth
The NY Times article implied that half of all cancers exhibit p53 mutations and the other half involve p53 inactivation through the binding of p53 to another cellular protein. Nutlins, drugs from Roche in Nutley NJ, and other drugs from Sanofi and Merck apparently can reverse the inactivation of p53 by driving a wedge between p53 and the protein bound inhibitor.
Alternatively, we think it’s worth proposing another drug target to reduce tumor growth based on the work of the Japanese team. A concise summary of the team’s results from luciferase reporter assays is below:
- TLS activates survivin transcription
- PRMT1, a protein arginine methlytransferase, activates survivin transcription
- TLS and PRMT1 synergistically co-activate transcription at the survivin promoter
- Catalytically-dead PRMT1 or treatment of cells with a methylation inhibitor both showed that the synergistic transcriptional activation is mediated by TLS arginine-methylation
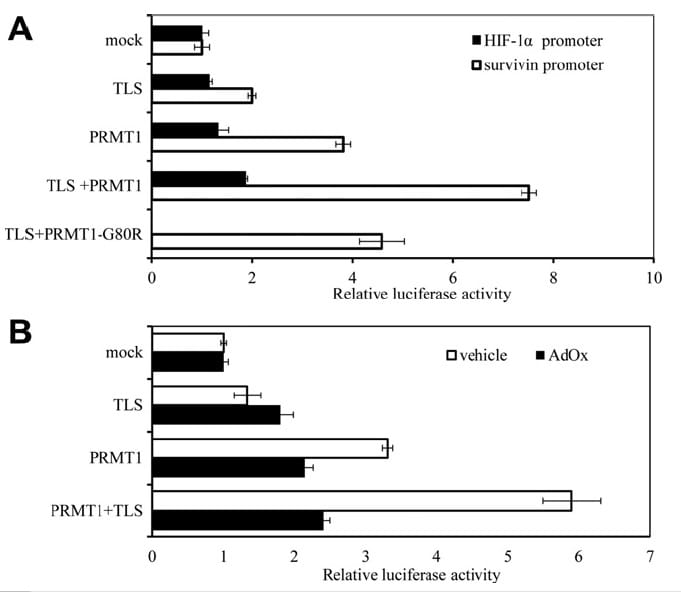
FIGURE 2. Synergistic TLS and PRMT1 co-activation of transcription at the survivin promoter
CONCLUSION: Arginine methylation of TLS produces more active transcription of survivin. Regardless of whether tumor growth is facilitated by mutation or
inactivation of p53, or by the TLS-CHOP oncoprotein, the most logical approach for broad drug effectiveness would be to focus on the final common step of survivin transcription. If pathologically high levels of survivin expression depend significantly on protein methylation, as suggested by the experiments reported above, a new comprehensive rationale for reducing tumor growth may be within reach.
By targeting protein arginine methyltransferase 1 (PRMT1) catalytic activity via treatment with inhibitors (Copeland et al., 2009), increased survivin expression can be reversed, tumor cell death re-activated and the size and viability of tumors will be reduced.
To make matters more complicated, of course, it should be pointed out that p53 is also regulated by arginine methylation (Jansson et al., 2008). In this case the methylation appears to be carried out by PRMT5, a fundamentally different type of arginine methyltransferase. We can try to sort that out later in future posts.
Send comments, criticisms, brain-storms and ideas about p53, survivin and the role of protein methylation in the biology of tumorigenesis to the CH3 News blog whether it is dogmatic, wild or just plain common sense.
Anybody know more about the Sanofi and Merck drugs?
All feedback is welcome!